Instrukcja wizualizacji struktur krystalicznych za pomocą DSV
(ćwiczenie z krystalochemii)
I. Cel ćwiczenia
Celem ćwiczenia jest nauka tworzenia modeli 3D budowy wewnętrznej kryształu, korzystanie z baz danych substancji krystalicznych i zapoznanie się z kilkoma przykładowymi strukturami.
Podstawą oceny wykonania ćwiczenia jest:
– obliczenie charakterów wiązań (oddziaływań) chemicznych;
– przedstawienie wiązań zgodnie z przyjętą konwencją;
– przedstawienie charakterystycznych podstruktur (np. łańcuchów).
II. Wstęp:
Na podstawie dostarczanych przez rentgenografię pozycji atomów w komórce elementarnej wiemy, które atomy (jony) sąsiadują ze sobą i jak się wzajemnie otaczają. Tym atomom szacujemy charakter oddziaływań. Na tej podstawie możemy zaklasyfikować tę substancję krystaliczną do właściwego jej typu struktur krystalochemicznych. Powielenie komórek pozwala zauważyć większe podstruktury, tworzą je zwykle fragmenty powiązane silniejszymi wiązaniami niż sąsiednie. Mogą to być fragmenty powiązane wiązaniami kowalencyjnymi lub jonowe wielościany koordynacyjne. Wraz z konwencją przedstawiania różnego typu wiązań i podstruktur umożliwia to czytelną wizualizację budowy wewnętrznej kryształu z uwzględnieniem natury występujących wiązań chemicznych.
Przypomnienie z poprzednich ćwiczeń:
– Elektroujemność a wiązanie chemiczne:
· Wiązanie kowalencyjne – charakteryzuje mała różnica elektroujemności sąsiadów i duża elektroujemność obu atomów.
· Wiązanie jonowe ––––––– charakteryzuje duża różnica elektroujemności sąsiadów i duża elektroujemność jednego z nich.
· Wiązanie metaliczne ––– charakteryzuje mała różnica elektroujemności sąsiadów i mała elektroujemność obu atomów.
– Szacowanie charakteru metalicznego jest utrudnione bo nie jest oczywiste na jakim stopniu utlenienia występują atomy w metalu. Przydatna jest wówczas odległość międzyatomowa znacznie wówczas większa od sumy typowych promieni jonowych.
– Główna różnica pomiędzy wiązaniem jonowym a kowalencyjnym to kierunkowość wiązania kowalencyjnego, zwykle jest to związane z obecnością pary elektronowej w przestrzeni między tymi atomami (reguła oktetu i inne), stąd np. atom siarki w siarczanach jest otaczany przez 4 atomy tlenu. W przypadku wiązania jonowego brak jest takiej zależności (np. atom sodu otacza 6 atomów chloru w krysztale NaCl mimo jednego tylko elektronu walencyjnego atomu sodu) i jony układają się jak zbiór naładowanych kulek (maksymalizując przyciągania a minimalizując odpychania) (tzw. Reguły Paulinga).
III. Narzędzia
Ćwiczenie przeznaczone jest do wykonania w Pracowni Komputerowej (B8, sala -1.24) za pomocą programu Discovery Studio Visualizer (DSV).
Pierwsza część ćwiczenia poświęcona jest na opanowanie podstawowych funkcji programu DSV na przykładzie tworzenia modeli 3D kilku wybranych struktur a w drugiej należało będzie samodzielnie zwizualizować dwie otrzymane struktury.
IV. Ogólny plan ćwiczenia
W celu wykonania ćwiczenia należy najpierw włączyć komputer i uruchomić program DSV. Po otrzymaniu nazwy substancji krystalicznej należy wykonać wstępną wizualizację (na razie niepoprawną) i na tej podstawie oraz przy pomocy tabeli elektroujemności wykonać odpowiednie obliczenia. Na podstawie tych obliczeń odpowiednio zmodyfikować wizualizację. Modyfikowanie należy zacząć od powielenia komórek w celu zorientowania się o obecności większych podstruktur. Poprzez zaznaczanie selektywne należy nadać wybranym atomom (jonom) czy podstrukturom odpowiedni wygląd. Gotowy model należy zgłosić do odbioru lub wysłać e–mailem a wypełnione formularze zwrócić.
V. Szczegółowy plan wykonania ćwiczenia
Kolejność działań w celu zwizualizowania struktury powinna wyglądać typowo tak:
Do pierwszego zadania należy:
1. Otworzyć plik !WZÓR.cif za pomocą Notatnika (Menu Start / Programy / Akcesoria / Notatnik) z kartoteki: Moje dokumenty \ Ćwiczenia z krystalochemii lub ściągnąć jego nie uszkodzoną wersję ze strony www.
2. Uzupełnić plik na podstawie otrzymanych danych o substancji chemicznej i na podstawie Międzynarodowych Tablic Krystalograficznych, Tom A (www.iucr.org lub awaryjnie z www.cryst.ehu.es). W tym celu należy w miejsca wygwiazdkowane (a nie gdzie indziej) wpisać grupę symetrii, brakujące parametry krystalograficzne i pozycję atomów.
Uwagi: Do pliku .cif wpisujemy ułamki dziesiętne a nie zwykłe. Np. 1/3 to 0.333 (i używamy kropki dziesiętnej a nie przecinka). Wystarczy podać pierwsze położenie spośród podanych w Tablicach położeń symetrycznie równoważnych („Wyckoff’a”) bo resztę program DSV wygeneruje automatycznie. Przy zapisywaniu Notatnikiem należy wpisać nazwę z końcówką .cif lub później włączyć w Windows pokazywanie końcówek plików i poprawić taki plik (bo niektóre komputery w Pracowni ukrywają końcówki plików i plik ma nazwę „nazwa.cif.txt” czego nie widać).
Do drugiego zadania należy plik ‘.cif’ ściągnąć z bazy danych. Uwaga, niektóre pliki ‘.cif’ wymagają otwierania w WordPadzie.
3. Tak stworzony (lub ściągnięty z bazy danych) plik zawiera komplet informacji potrzebnych do odtworzenia struktury krystalicznej i należy go zapisać w tej samej kartotece pod nazwą: Grupa_Imię_Nazwisko.cif
(uwaga, przy zapisywaniu „Notatnikiem” należy użyć formatu ANSI a nie UTF-8);
4. Za pomocą programu DSV „Discovery Studio Visualizer” (MenuStart / Programy / Accelrys / DS Visualizer) wczytać zapisany wcześniej plik ‘.cif ‘
5. Ocenić poprawność wczytanego pliku ‘.cif’ (np. brak ponakładanych lub za blisko leżących atomów, ewentualnie zbyt duże pustki, odstępy międzyatomowe). Jeśli pojawią się błędy należy powrócić do Notatnika (lub WordPada) w celu dokonania korekty lub próbować zmieniać tzw. origin w DSV, inną przyczyną może być użycie niewłaściwej wersji pozycji Wyckoffa (grupy w Tablicach mają różne wersje różniące się wyborem osi lub punktu zerowego). Jeśli wersja grupy jest zanotowana w pliku ‘.cif’ jako np.: 'F d d d :2' to należy ta skrócić do 'F d d d' i potem w DSV ręcznie zmienić ‘origin’.
6. Przez powielenie komórek należy określić czy i jakie aniony złożone, pierścienie, łańcuchy, warstwy i komory występują w strukturze. Struktury te wyróżnia połączenie silniejszym wiązaniem. Inną cechą możliwą do zaobserwowania przez powielenie komórek jest obecność ciągłej więźby wiązań kowalencyjnych. Uwaga praktyczna: najpierw zdecydować ile potrzeba komórek a potem dopiero zmieniać ‘Display Style’ a jeśli struktura będzie zawierać wielościany to zacząć od ‘Display Style – Polyhedron’.
7. Na podstawie wyświetlanej na ekranie struktury i wzoru chemicznego tej substancji określić jakiego typu wiązania występują w wybranej strukturze. W tym celu należy policzyć jonowość wiązań oraz wytrzymałości wiązań (na podstawie odczytanych z ekranu liczb koordynacyjnych kationów i ich stopni utlenienia). Stopnie utlenienia wyliczamy na podstawie wzoru sumarycznego, elektroujemność odczytujemy z załączonej tablicy (z elektroujemnościami Görlicha) a liczbę koordynacyjną odczytujemy z ekranu (oczywiście po prawidłowym zbudowaniu modelu). (Liczba koordynacyjna to parametr dotyczący struktur jonowych i określa ilość najbliższych równo odległych sąsiadów no i jak to w strukturach jonowych, są oni oczywiście przeciwnego znaku). Dla wszystkich par sąsiadów, dla których %I > 40 liczymy wytrzymałość wiązania kation–anion: WK-A = Stopień utlenienia / Liczba koordynacyjna. Jeśli jest ona większa niż połowa ładunku anionu to ten kation wraz z otaczającymi go anionami prostymi tworzy anion złożony (typowym przykładem jest np. SO4-2).
8. Konieczne jest skorygowanie sposobu wyświetlania zapronowanego przez DSV, zwykle niektóre wiązania są niepotrzebne. Zmiana trybu wyświetlania wybranych atomów na CPK jednocześnie likwiduje wiązania, ale nie zawsze tak się dzieje, więc czasem należy ręcznie skasować wiązania pomiędzy jonami, wiązania do atomów z drugiej strefy koordynacji i inne zbędne. (Żeby je skasować wystarczy je zaznaczyć i nacisnąć klawisz Del. Jest to ważne, bo program DSV tworzy wielościany tam, gdzie sięgały wiązania, więc nadmiarowe wiązania powodują powstanie wielościanów nieforemnych, pozbawionych sensu chemicznego. Analogicznie, w celu utworzenia wielościanu tam gdzie nie chce się utworzyć automatycznie należy dorysować wiązania (ikonka: żółty ołówek).
9. Dobrać sposób wizualizacji struktury do charakteru występujących w niej wiązań chemicznych. W tym celu należy najpierw zdecydować, które atomy jak przedstawiamy. Czyli gdzie są jony, gdzie wiązania kowalencyjne, gdzie pierścienie, czy jest ciągła więźba wiązań kowalencyjnych itp. Atomy, którym zamierzamy zmienić sposób wyświetlania najpierw należy selektywnie zaznaczyć. A następnie wykorzystać możliwości oferowane przez program DSV. Np:
– wybrać model czaszowy, wielościanowy, szkieletowy lub mieszany (według załączonej tabeli);
– wybrać więcej niż jedną komórkę elementarną (dla pokazania np. pierścieni, ciągłej więźby…);
– ustawić jonom odpowiednie do stopnia utlenienia promienie jonowe (według załączonej tabeli);
Anion złożony przedstawiamy typowo wielościanem w celu odróżnienia od jonów prostych. Jeśli przedstawienie anionu złożonego w postaci wielościanu nie jest możliwe (np. dla CO3-2) to pozostaje przedstawić go w postaci przecinających się kul. Można (ale niekoniecznie) tak samo przedstawić strukturę gdy nie ma anionów złożonych ale kationy znacznie różnią się między sobą (np. jeden duży i o dużej LK a drugi mały i o małej LK). Jeśli w strukturze występują dwie podsieci, ale o bardzo podobnej wytrzymałości wiązań, (np. 4/4 i 3/4 lub 5/4) to obie należy wyróżnić wielościanami.
10. Gotowy model zapisać na dysku w formacie .dsv i .png i zabrać się za robienie drugiego. Gotowe modele (lub pierwszy model) należy zgłosić do odbioru. Jeśli czas nie pozwoli na odbiór wszystkich prac w trakcie zajęć to należy wysłać pod koniec zajęć (ale nie po) na adres email TRZY pliki: kopię ekranu (printscreen lub Save As .png ), plik .cif i plik .dsv. (Kopia ekranu jest konieczna ponieważ w pliku .dsv nie przenoszą się promienie jonowe i struktura zmienia wygląd.)
VI. Konwencja wizualizacji przyjęta w celu wykonania niniejszego ćwiczenia
(Nie ma w chemii jednego obowiązującego standardu przedstawiania atomów, jonów i wiązań chemicznych ale na potrzeby niniejszych ćwiczeń przyjmujemy dosyć sensowną i popularną konwencję.)
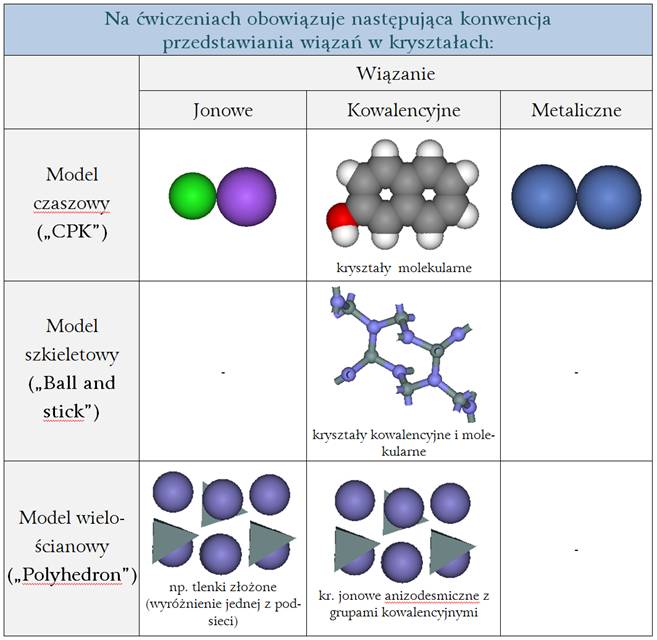
Znaczenie używanych elementów:
– Niestykające się kule modelu czaszowego („CPK”) = brak wiązania lub wiąz. Van der Waalsa;
– Stykające się kule modelu czaszowego („CPK”) = wiązanie jonowe lub metaliczne;
– Przecinające się kule modelu czaszowego („CPK”) = wiązanie kowalencyjne;
– Model szkieletowy („Stick”, „Ball and stick”) = wiązanie kowalencyjne;
– Model wielościanowy („Polyhedron”)
= kompleks atomów (jonów) połączony wiązaniami
kowalencyjnymi lub wielościan koordynacyjny w strukturze jonowej (dla
wyróżnienia którejś
z podsieci). Proszę zwrócić uwagę, że np. w przypadku wielościanów tlenowych
tleny można
„wyłączyć”, bo sam wielościan już je pokazuje.
VII. Odbiór pracy
Obrazek powinien ilustrować naturę występujących wiązań chemicznych. Gotowa praca powinna wyglądać np. tak:
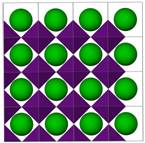
Ba(BiO3)
Elementy oceniane i punktacja wyszczególnione są na dostarczonym formularzu. W szczególności przed zgłoszeniem pracy należy zwrócić na:
· czy na liście wiązań są wymienione wszystkie pary sąsiadów występujące w strukturze i nie ma takich par, które ze sobą nie sąsiadują;
· czy we wnioskach na formularzu skreślono wszystkie nieprawdziwe sformułowania;
· czy struktura wygląda sensownie, np. cy atomy nie leżą jeden prawie na drugim, nie ma wielościanów nieforemnych, nie ma wiązań do atomów z drugiej strefy koordynacji, nie ma wiązań tam gdzie są oddziaływania jonowe itp. i czy zapisano wynik na dysku jako .msv i .png;
· czy jest odpowiednia ilość komórek dla pokazania większych struktur (np. warstw, ciągłej więźby):
(Pierścienie to zwykle 3x3x2 komórki, łańcuchy to zwykle 7x3x2 komórki, warstwy to zwykle 7x7x2 komórki, izolowane aniony złożone (wielościany lub przecinające się kule dla płaskich) to zwykle 3x3x3 komórki. Ciągła więźba wiązań kowalencyjnych to zwykle 4x4x4 komórki.)
· czy podsieć o większej wytrzymałości wiązań (lub dwie podsieci o podobnej sile wiązań) jest wyróżniona wielościanami (lub w inny sposób);
· czy atomy w narożach wielościanów są małymi kuleczkami lub są w ogóle wyłączone;
· czy jony mają ustawione odpowiednie promienie jonowe i czy dla nich włączony jest tryb CPK (bo tylko w tym trybie używane są promienie jonowe) i czy skala CPK=1;
· czy kule jonów nie przecinają się;
· czy tam gdzie jony sąsiadują to się stykają (za wyjątkiem tych z naroży wielościanów);
· czy wszystkie atomy (występujące w narożach tetraedrów) danego typu należało wyłączyć (np. czasami jedne atomy tlenu tworzą wielościan wokół kationu a drugie tworzą grupy OH lub warstwę tlenkową – należy je wówczas ręcznie po jednym zamienić na CPK);
· czy nie zapomniano o obojętnych molekułach (np. H2O, NH3), które zwykle przedstawia się modelem czaszowym;
· czy wybrano taką orientację modelu 3D, na której widać jej cechy charakterystyczne.
VIII. O instrukcji obsługi programu DSV
Program DSV jest opisany w jego menu (Menu->Help) i dublowanie tu tego opisu byłoby niepotrzebną pracą. Do wykonania ćwiczenia potrzebny jest tylko niewielki podzbiór jego możliwości. Pierwsza część ćwiczeń jest właśnie instruktażem jak korzystać z programu. Doszlifowanie tej umiejętności proszę potraktować, jako zadanie domowe. Poniżej zebrano trochę uwag dotyczących Obsługi DSV przydatnych do wykonania ćwiczenia:
Przydatne uwagi:
– Najpotrzebniejsze opcje:
|
|
View/Display Style |
-przełącza
sposoby wizualizacji |
|
– |
Structure/Crystal Cell/Edit Parameters |
-powielanie komórki elementarnej |
|
|
Tools/Fit to Screen |
-narzędzie do centrowania obrazu |
|
|
– |
-tryb myszy (zaznaczanie/obracanie) |
|
– |
Edit/Select |
-selektywne zaznaczanie atomów |
|
– |
Chemistry/Element Properties |
- ustawianie promieni jonowych |
– Program DSV
musi mieć odznaczone opcje: Edit->Preferences->Molecules->Import->Create_animation.
Innym problemem jest wartość: Structure/Crystal
Cell/Edit Parameters/Preferences/Special Positions Tolerance = 0 -> należy wpisać np. 0.05. Ma to związek z dokładnością podania
pozycji atomów w pliku ‘.cif’. Jeżeli dwa atomy są bliżej siebie niż ta tolerancja to
zostaną uznane za ten sam atom.
– Kolory, które program domyślnie stosuje dla rozróżnienia atomów, traktujemy, jako umowną konwencję obowiązującą na niniejszych ćwiczeniach i na wykładzie – nie należy ich zmieniać.
– Uwaga na zaznaczenie atomu lub wiązania na żółto (przez kliknięcie na nim), bo wówczas narzędzia nie działają na całą strukturę a tylko na zaznaczenie a niektóre narzędzia są wówczas nieaktywne. Nawet centrowanie obrazu to uwzględnia. Przez zaznaczenie wybranej grupy atomów (Edit/Select…) można część atomów wyświetlić inaczej niż pozostałe.
– Zwykle dobrze jest zaraz po pierwszym wczytaniu .cif włączyć: Structure/Crystal Cell/Edit Parameters/Preferences/Symmetry Style: One Cell – powoduje to translację atomów o ujemnych położeniach w pozycji Wyckoff’a do tej komórki w której wypadają dodatnie.
– W przypadku używania trybu CPK należy wpisać każdemu występującemu rodzajowi atomów odpowiedni promień jonowy (Chemistry/Element Properties...) i pozostawić skalę CPK =1.0 (View/Display Style/CPK Scale…).
– Wygodnym trybem do policzenia najbliższych sąsiadów (gdy nie da się tego zrobić zliczając wiązania) jest tryb CPK (często ze zmniejszoną skalą, np. CPK=0.7).
– Dla osób zainteresowanych modelowaniem i oglądaniem struktur na własnym komputerze program DSV dostępny jest (po rejestracji) na stronie: http://accelrys.com/products/discovery-studio/visualization-download.php W celu skonfigurowania go tak jak na niniejszych ćwiczeniach należy w Edit/Preferences zaznaczyć: 3DWindow/Import/Display Unit Cell i odznaczyć: Convert 2D molecules. Przydatne jest też zaznaczyć Enable Hardware Acceleration i Display Styles /Ball&Sticks oraz podniesienie jakości grafiki: 3DWindow/Quality/High i odznaczyć opcję Edit -> Preferences -> Molecules -> Import -> Create_animation
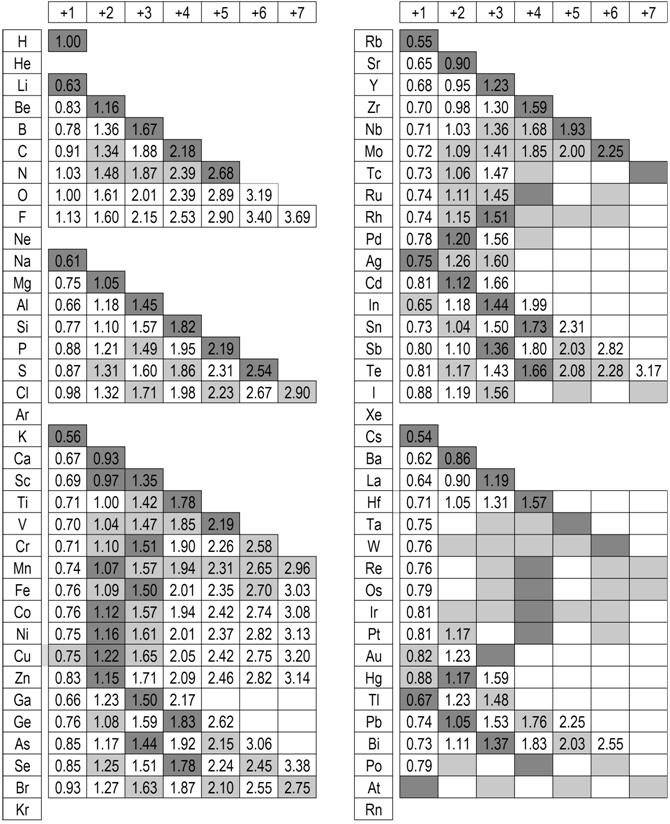
XI. Elektroujemności jonów według skali Görlicha
Dla wygody dołączono tu (z podręcznika) tabelę elektroujemności w skali Görlicha. Tabela ta uwzględnia zależność elektroujemności jonu od jego stopnia utlenienia według teorii elektroujemności Görlicha. Według tej teorii elektroujemność jest ładunkiem efektywnym liczonym, jako pierwiastek z odpowiedniego potencjału jonizacji wyrażonego w rydbergach. Elektroujemność dla jonu o ładunku zrębu atomowego jest elektroujemnością pierwiastka (jest jego cechą tak jak np. kolor dla pierwiastka Cu). Dzięki tak zdefiniowanej elektroujemności można obliczyć spodziewany procent jonowości wiązania (oddziaływania) dwóch jonów za pomocą wzoru: %I=(1–xG/x’G)*100% gdzie (xG<x’G). Dla kationów używa się xG dla odpowiedniego stopnia utlenienia. Dla anionów używa się xG z kolumny odpowiadającej ładunkowi zrębu atomowego.
Wyjaśnienie: Nie oznacza to, że traktujemy anion tak jakby był na takim dziwnym stopniu utlenienia. Jest to sposób liczenia pasujący do podanego wyżej wzoru (model Görlicha). Według tego modelu zakładamy początkowo, że nie wiemy jak się przesunie wspólna chmura elektronów walencyjnych. Zakładamy również, że działa na nie z jednej strony kation, (czyli zrąb (rdzeń) atomowy wraz z nieoderwanymi elektronami walencyjnymi, jeśli takie są) a z drugiej zrąb atomowy anionu). Np. w Fe2O3 będzie to Fe+3 i O+6. Dopiero w wyniku obliczenia %I wiemy jak przesuną się wspólne elektrony walencyjne czyli szacujemy spodziewaną polaryzację wiązania. Takie liczenie nie uwzględnia wpływu otoczenia, jest więc jedynie pierwszą wskazówką (potrzeba jeszcze policzyć WK-A). Wcześniej należy oczywiście policzyć stopnie utlenienia atomów we wzorze sumarycznym związku chemicznego.
Ciemniejszym tłem zaznaczono typowe stopnie utlenienia.
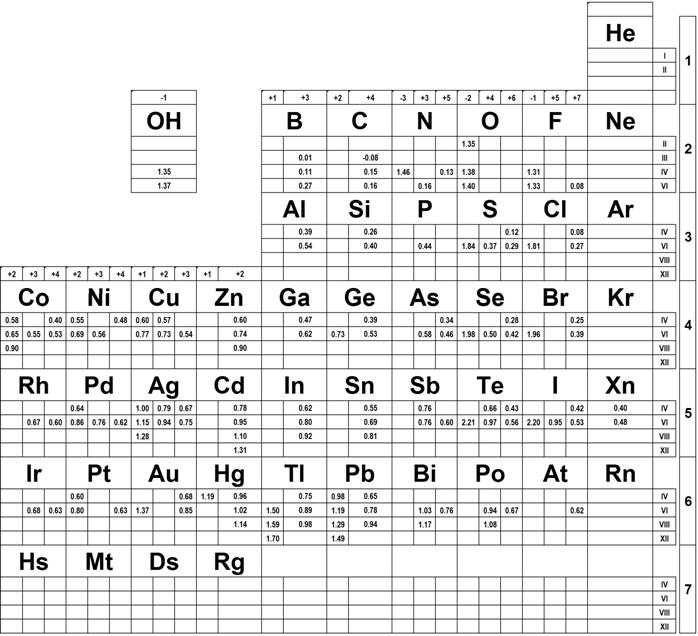
X. Tabela promieni jonowych
Dla wygody dołączono tu poniżej tabelę (z podręcznika) promieni jonowych wg Shanona i Prewita. Tabela ta uwzględnia zależność promienia jonowego od stopnia utlenienia i liczby koordynacyjnej.
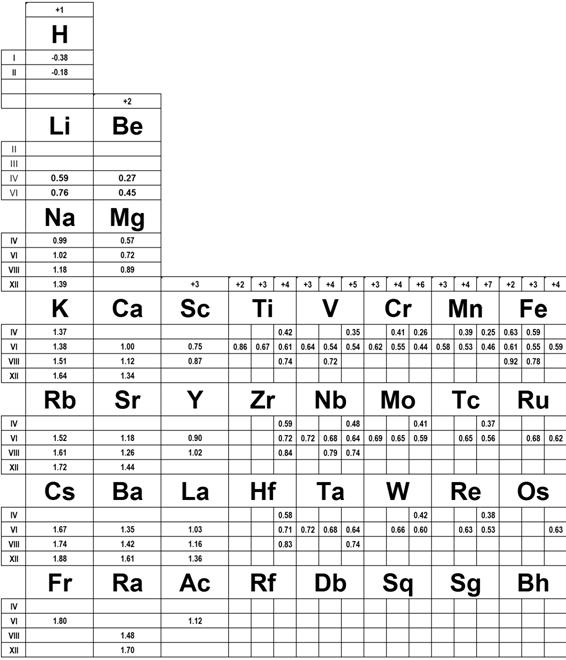
Cyframi rzymskimi z prawej i lewej rozróżniono liczby koordynacyjne a arabskimi u góry stopnie utlenienia